N体問題
N体問題は重力多体問題や分子動力学などの物理現象自体が離散的な点の相互作用で記述される分野で盛んに用いられてきた。しかし、N体問題の解法は弾性力学、流体力学、電磁気学、音響学、量子力学などの数値解析にも用いることができる。これは連続体を記述する偏微分方程式を積分方程式に変換し求積点を用いて積分を行うことで離散点同士の相互作用問題に帰着するためである。そのため、N体問題の高速解法は科学技術計算において欠かすことのできないアルゴリズムであるといえる。
高速多重極展開法
高速多重極展開法(FMM)はN体問題の計算コストをO(N2)からO(N)に軽減する手法である。FMMはFFTやKrylov部分空間法などと並んで20世紀の10大アルゴリズムの一つと考えられている。通常Byte/flopの低い蜜行列などのアルゴリズムはその複雑性が高くO(N3)、複雑性の低いFFTや疎行列計算などのアルゴリズムはByte/flopが高い。しかし、FMMはO(N)の複雑性も有しながらも蜜行列積よりも低いByte/flopを実現できる。つまり、FMMは効率的なアルゴリズムでありながらも演算密度が高く、次世代の計算機アーキテクチャにおける様々な楕円型方程式の高速解法の代替手法として期待されている。
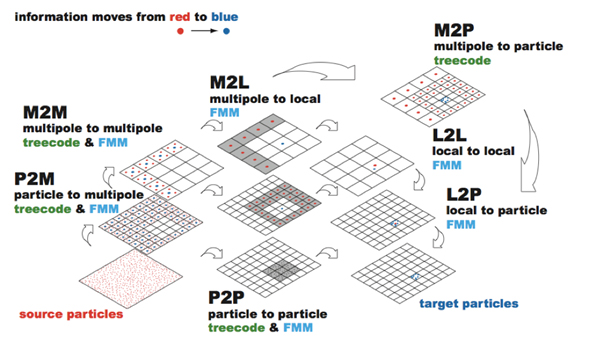
図1 FMMの計算の流れ
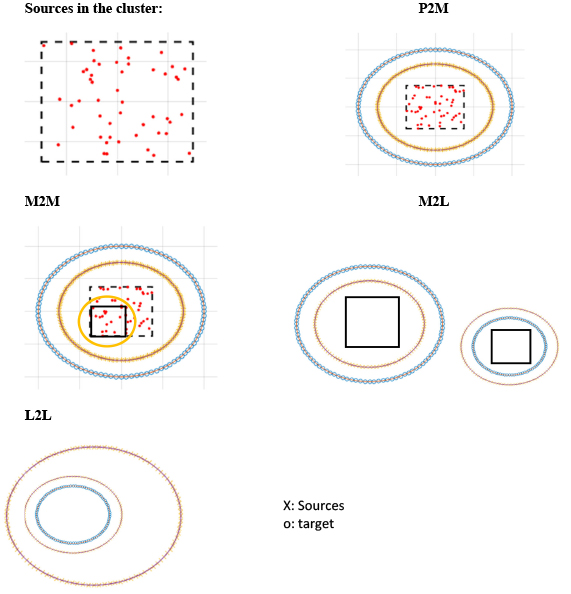
N体問題の計算を高速化する手法としてFMMは非常に有効である。FMMは6つの部分から成り立っている。P2Mは粒子の情報から多重極展開への変換、M2Mは多重極展開から多重極展開での変換、M2Lは多重極展開から局所展開への変換、L2Lは局所展開から局所展開への変換、L2Pは局所展開から粒子の作用への変換、P2Pは粒子同士の直接相互作用を表す。また、treecodeはFMMと異なり、M2L、L2L、L2Pの代わりにM2Pの変換を行うことで多重極展開から粒子での直接の作用を計算する。
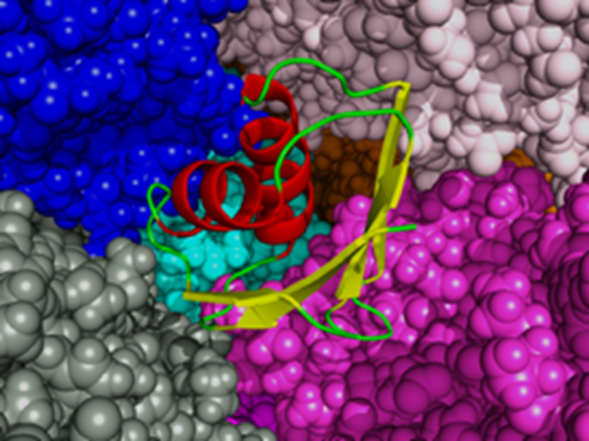
図2 タンパク質のクラウディングの分子シミュレーション
分子動力学による解析は一般的に水で満たされた周期境界条件のような理想化された試験環境下(in vitro)で行われる。一方、実際の細胞などは様々なタンパク質が混在しており、そのような環境下での挙動をin vitroの計算から予測するには限界がある。本研究では663,552コアを用いた並列計算で5億原子の分子シミュレーションを行い、タンパク質のクラウディングの影響を調べた。
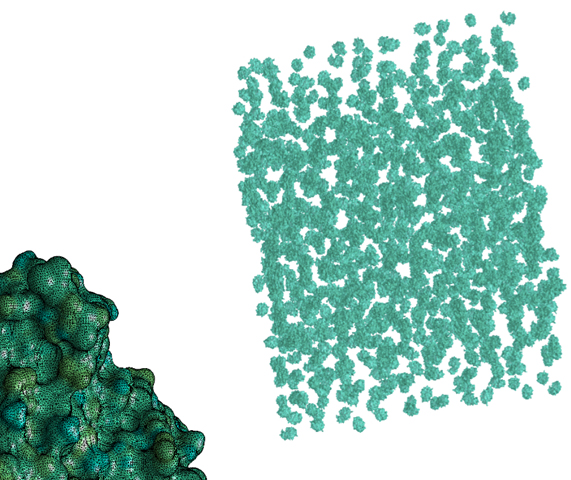
図3 境界要素法を用いた暗溶媒による分子シミュレーション
分子シミュレーションは水分子を陽的に扱う陽溶媒と、連続体として扱う暗溶媒がある。暗溶媒の場合は分子の表面を境界要素法で離散化し、その上で積分方程式を解くことでポテンシャル場や力場の解析を行う。図3に示すように大規模解析においては多数の分子の集まりを境界要素法でそれぞれ離散化し、まとめて解くことで分子が混在するような場を解析することができる。
図4 渦法を用いた一様等方性乱流の解析
乱流解析も渦を最小単位とするような離散化を行うことでN体問題として解くことができる。これにより、FMMを用いてPoisson方程式に相当する部分をO(N)の計算時間で解くことができ、大規模並列計算において良いスケーリングを得ることができる。
Implementation of Kernel Independent Fast Multipole Method (KIFMM)
KIFMM is a technique that was developed to speed up the calculation of long-ranged forces in the n-body problem from O(N2) to O(N). It doesn’t requires expanding the system green’s function using a multipole expansion like the classical FMM and it is based only on kernel evaluations.
Instead of using analytic expansions to represent the potential generated by sources inside a box of the hierarchical FMM tree like classical FMM, we use a continuous distribution of an equivalent density on a surface enclosing the box. To find this equivalent density we match its potential to the potential of the original sources at a surface, in the far field, by solving local Dirichlet-type boundary value problems. The far field evaluations are sparsified with singular value decomposition in 2D or fast Fourier transforms in 3D. Its advantage is the (relative) simplicity of the implementation.
Currently I am working on implementing a GPU scalable version of KIFMM.
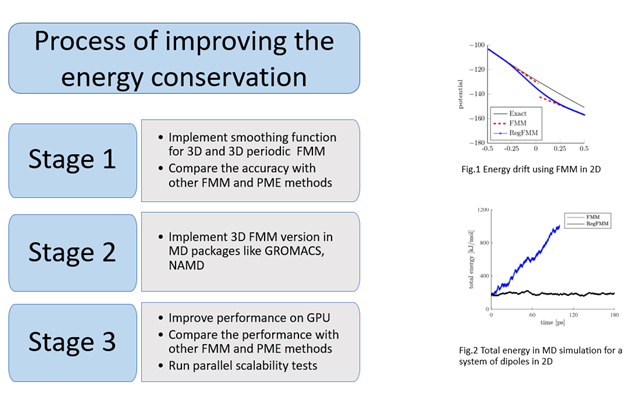
https://github.com/exafmm/exafmm-t/tree/gpuImproving the Energy Conservation of the FMM for Molecular Dynamics Simulations
Molecular dynamics simulation is common technique for studying the movements of atoms and molecules and can be described as a type of N-body system, a problem that can be solved by Fast Multipole Method. FMM is one of the fastest algorithms and it decreases the computational cost to O(N). Although FMM is considered to be a of a great importance in MD simulations, often during the simulation it can be observed a large energy drift caused by jump in the potential when particles move from one cell to another in the FMM hierarchical tree. The energy conservation is one of the parameters commonly used to describe the stability of MD simulation and the purpose of this research is implementing a regularization technique that will reduce the energy drift by smoothing the potential and obtain good energy conservation of the FMM for MD simulations.